Notes on DFT hands-on session of MSE6701H-CHN¶
约 2190 个字 153 行代码 27 张图片 预计阅读时间 9 分钟
Content: [TOC]
0.0 Notice¶
同学们好,今晚的课程为 DFT 部分上机实操课:
-
请同学们尽量都准备好笔记本电脑并充保持充足的电量(充电器也可以带上),条件允许的同学可以把排插带上。
-
本次上机实操课将涉及 DFT 性质计算(如晶格常数、能带、键长、空位形成能、表面能等)及 VASP 实操(如收敛性测试、静态计算、弛豫计算、能带计算、态密度计算等)等内容。
-
不同性质的计算,VASP 输入文件相关参数会有所不同,查阅官网手册了解参数设置含义,体会 “some practical concerns - First principles calculations within DFT” 课程内容的关联。
-
在使用课程材料前,请先阅读该目录下的
README文件内容来大致了解其使用说明、框架及功能,不要一开始就 “盲目” 进行操作(提交任务),以免出现不必要的错误。 -
使用课程材料过程中,请熟悉课程材料的目录及文件结构!课程材料及该 Notes 均测试正常。
0.1 Helpful things¶
可能有帮助的一些教程链接:
-
《多尺度材料模拟与计算》课程 MD、DFT 部分实验材料 Gitee 仓库
-
DFT 课程材料使用指南
-
VASP INCAR 参数:Category:INCAR tag - Vaspwiki
-
VASP POSCAR:POSCAR - Vaspwiki
-
VASP KPOINTS:KPOINTS - Vaspwiki
-
VASP 赝势推荐:Available PAW potentials - Vaspwiki
-
VASPKIT Features: Features — VASPKIT 1.5 documentation
-
模型构建相关开源程序
1. Preparation¶
1.1. Access to the cluster¶
We are still going to use Siyuan cluster for the hands-on session for the DFT part. Your temporary account should still be working.
If you forget how to access the Siyuan cluster, please refer to the manual for the MD part.
1.2. Update MMMS Course Materials¶
- Update(
git pull) the latest MMMS Course Materials
- or
2. Step by step instructions on the DFT examples¶
2.1. Single point calculation for FCC Cu¶
2.1.1. Without convergence test¶
- Get into the first example
- it should look like:
- We'll first try to carry out the calculation without convergence test. Get into the corresponding directory
- directory structure:
:::danger
Please check the content of all the files to have an idea on the calculation. Especially the README and job.slurm:
:::
- Run the calculation
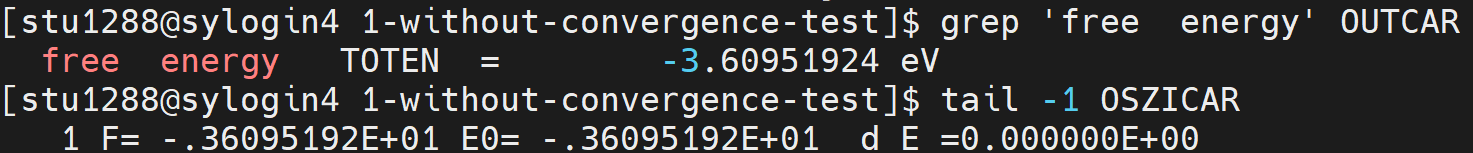
- Once the job is completed, you can check the results(Total energy) in
OUTCARorOSZICAR.
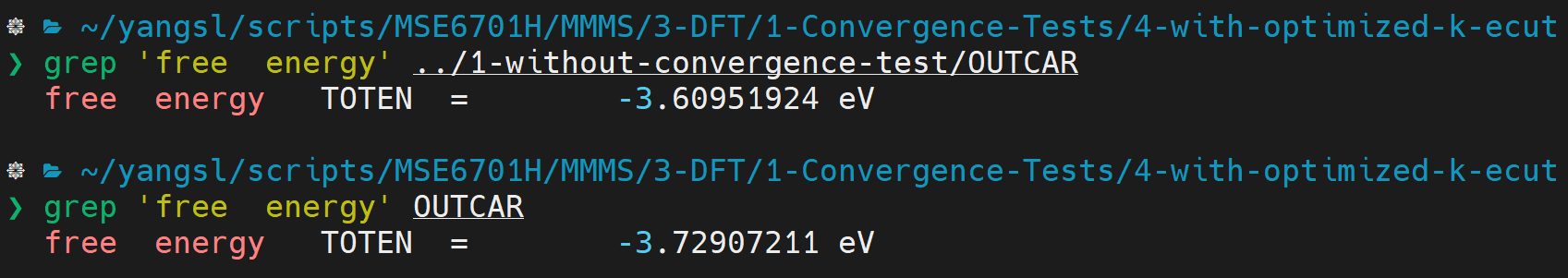
:::danger NOTE: there are exactly two spaces between 'free' and 'energy'. :::
- (Optional) Get the total k-points
2.1.2. Convergence test with respect to k-mesh size¶
- Get into the directory
:::danger
Please check the content of all the files to have an idea on the calculation. Especially the README and job.slurm:
:::
- Run your job
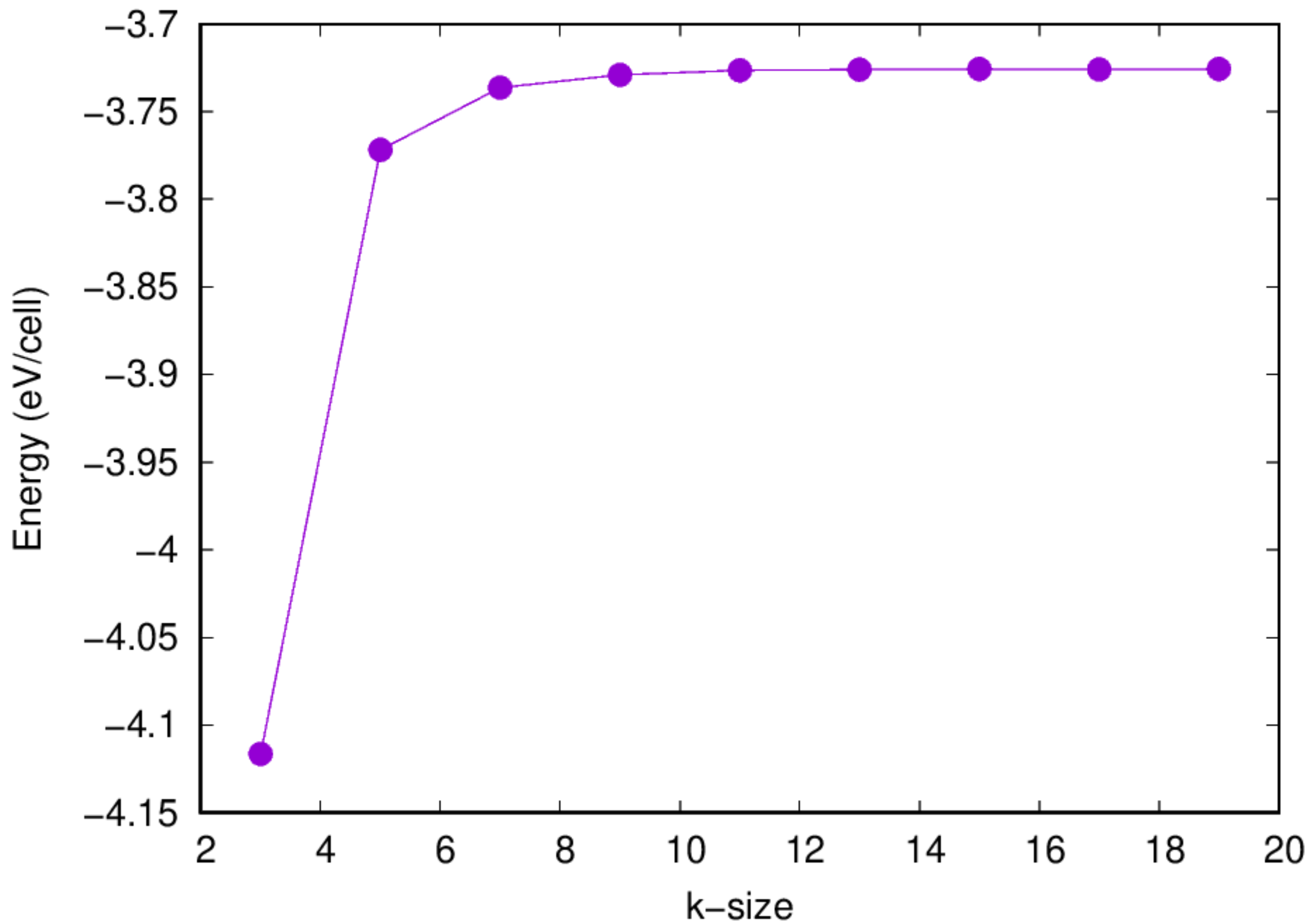
- Once completed, check the
eng_k.datfile for the convergence information
- And you can use the
check_conv_kscript prepared to visualize the convergence information:
- A figure should be shown to visualize the convergence information.
2.1.3. Convergence test with respect to plane wave cutoff energy¶
- Get into the directory
:::danger
Please check the content of all the files to have an idea on the calculation. Especially the README and job.slurm:
:::
- Run the calculations, it might take some time
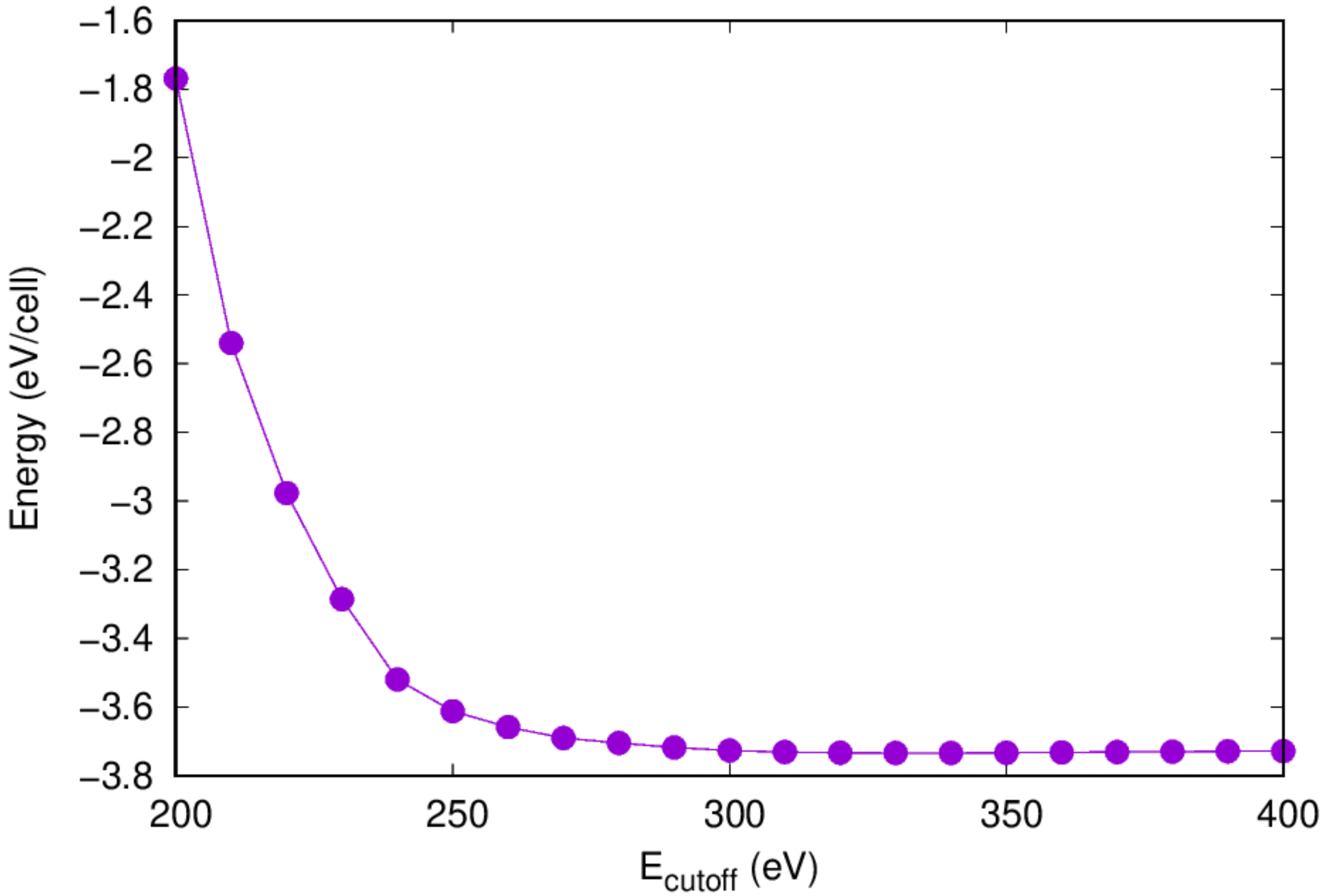
- Once the calculation is completed, check the
eng_ecut.datfile for the convergence information
- You can also use the
check_conv_ecutscript prepared to visualize the convergence information:
- A figure should be shown to visualize the convergence information.
With convergence test¶
- Get into the directory
- Run the calculation
- Once completed, check the
OUTCARfor the information. Compare the energy to that without convergence test
2.2. Lattice constant calculation¶
2.2.1. FCC Cu¶
- Get into the directory
:::danger
Please read through all the main input files of vasp, especially INCAR
:::
- Check the scripts
- Run the calculation
- Find the equilibrium lattice constant: Once the job is completed, you should find a file named
eng_a.datwith contents under your directory, you can fit the data to an Equation of States and obtain the equilibrium lattice constant by:
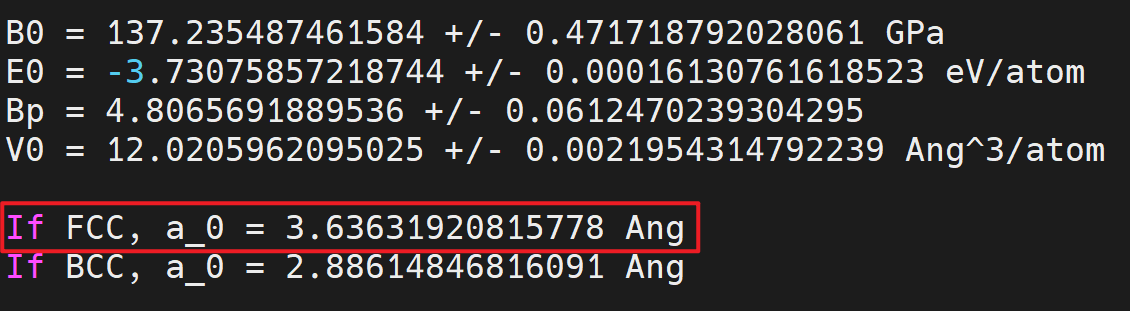
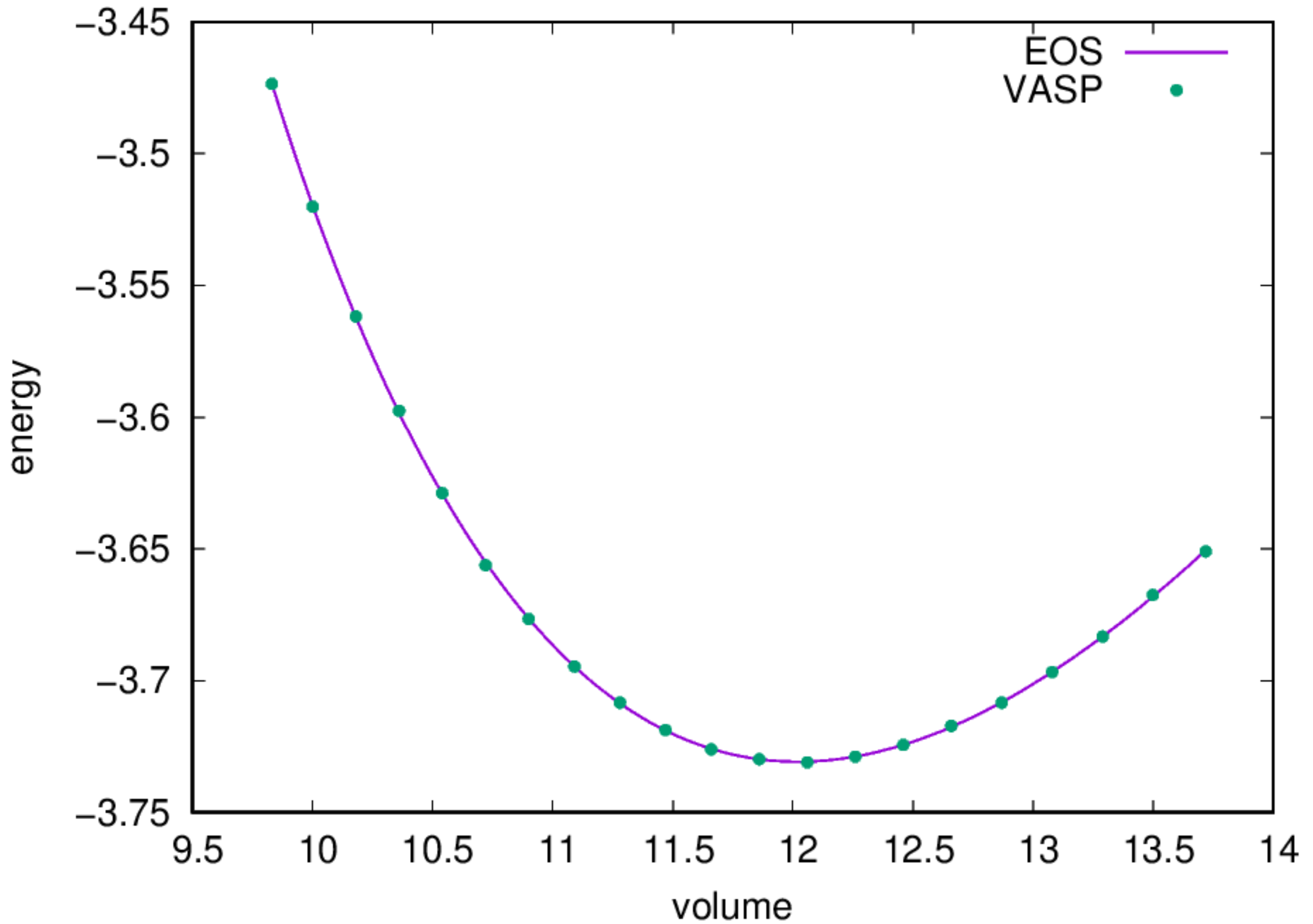
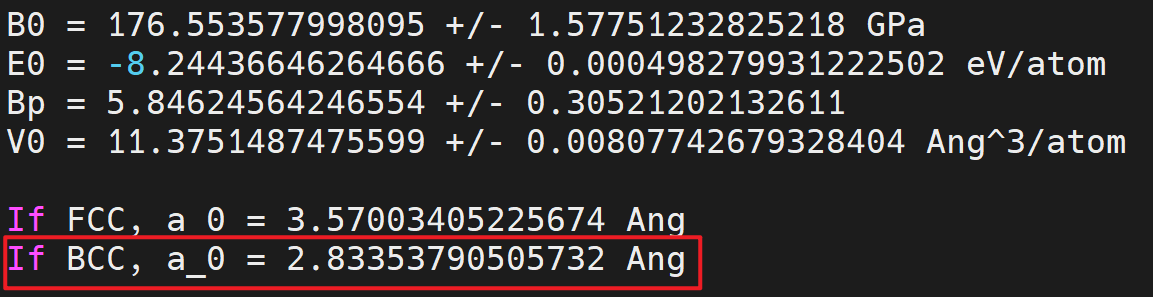
2.2.2. BCC Fe¶
- Get into the directory
:::danger
Please read through all the main input files of vasp, especially INCAR
:::
- Check the scripts
- Run the calculation
- Find the equilibrium lattice constant
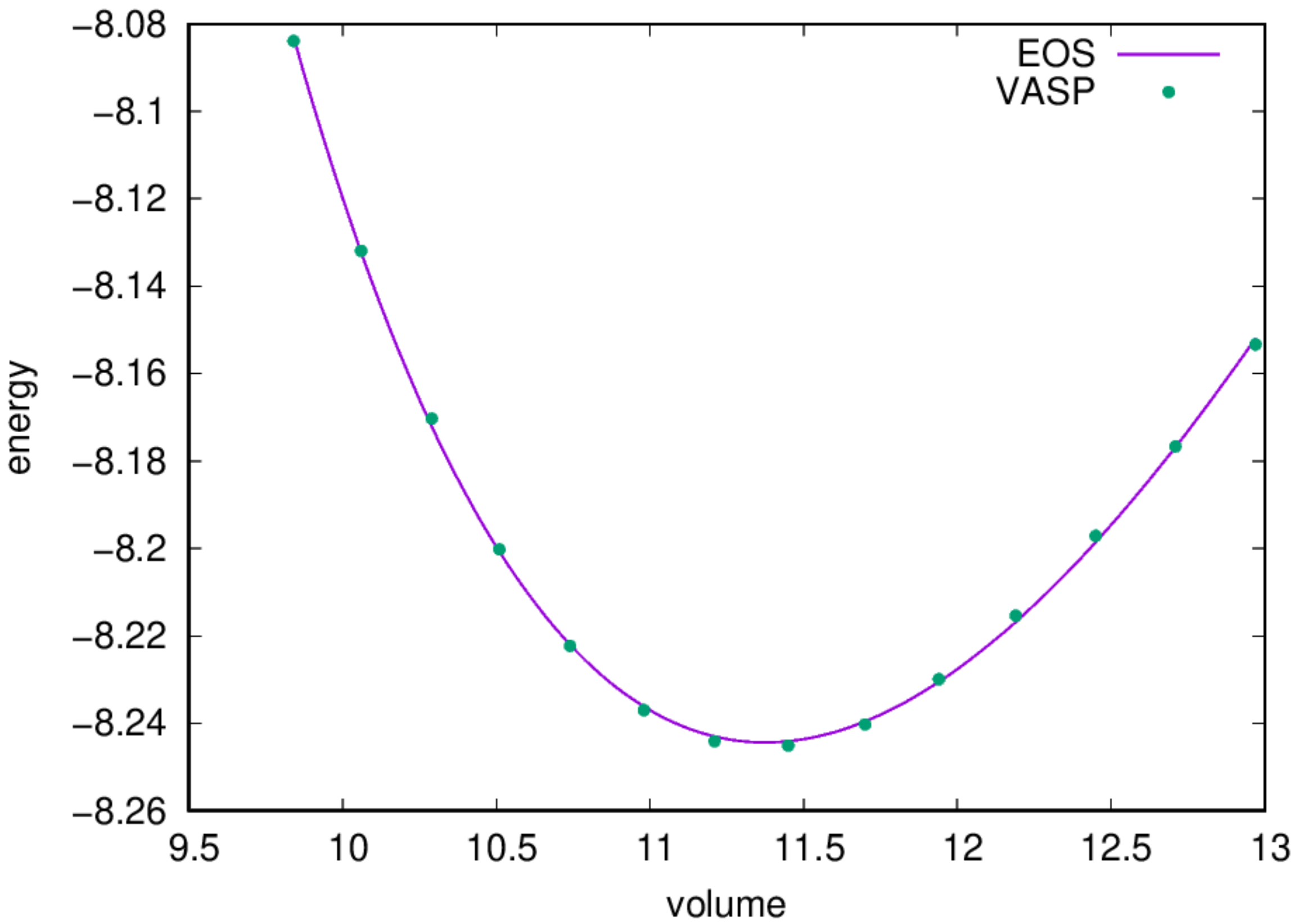
2.3. Band structure Calculations¶
For band structure calculations, generally one should perform two (or three if DOS is also wanted) calculations in sequence:
-
Self-consistent calculation. By running a usual single point energy calculation on a uniform k-mesh to obtain the ground state electron density.
-
Non-self-consistent calculation of band structure. By fixing the electron density as obtained from the previous run (ICHARG = 11 in
INCAR), the eigenvalues for different bands along a prescribed k-path will be calculated. Since in this case the k-path is not a uniform sampling of the reciprocal space. -
Non-self-consistent calculation of density of states. Generally this step is not quite necessary, but sometime useful. One also keeps the electron density from the first step fixed (ICHARG = 11 in
INCAR), but performs a calculation on a much denser k-mesh than the first step to obtain a smooth electron density of states.
2.3.1. FCC Cu¶
- Get into the directory
:::danger
Please read through all the main input files of vasp, especially INCARs and KPOINTSs. The three INCARs and three KPOINTSs correspond to the scf and nscf calculations, respectively.
:::
- Check the scripts
-
Run the calculation
-
Examine the band structure and DOS: Once the job is completed, you should be able to get the band structure via the following command:
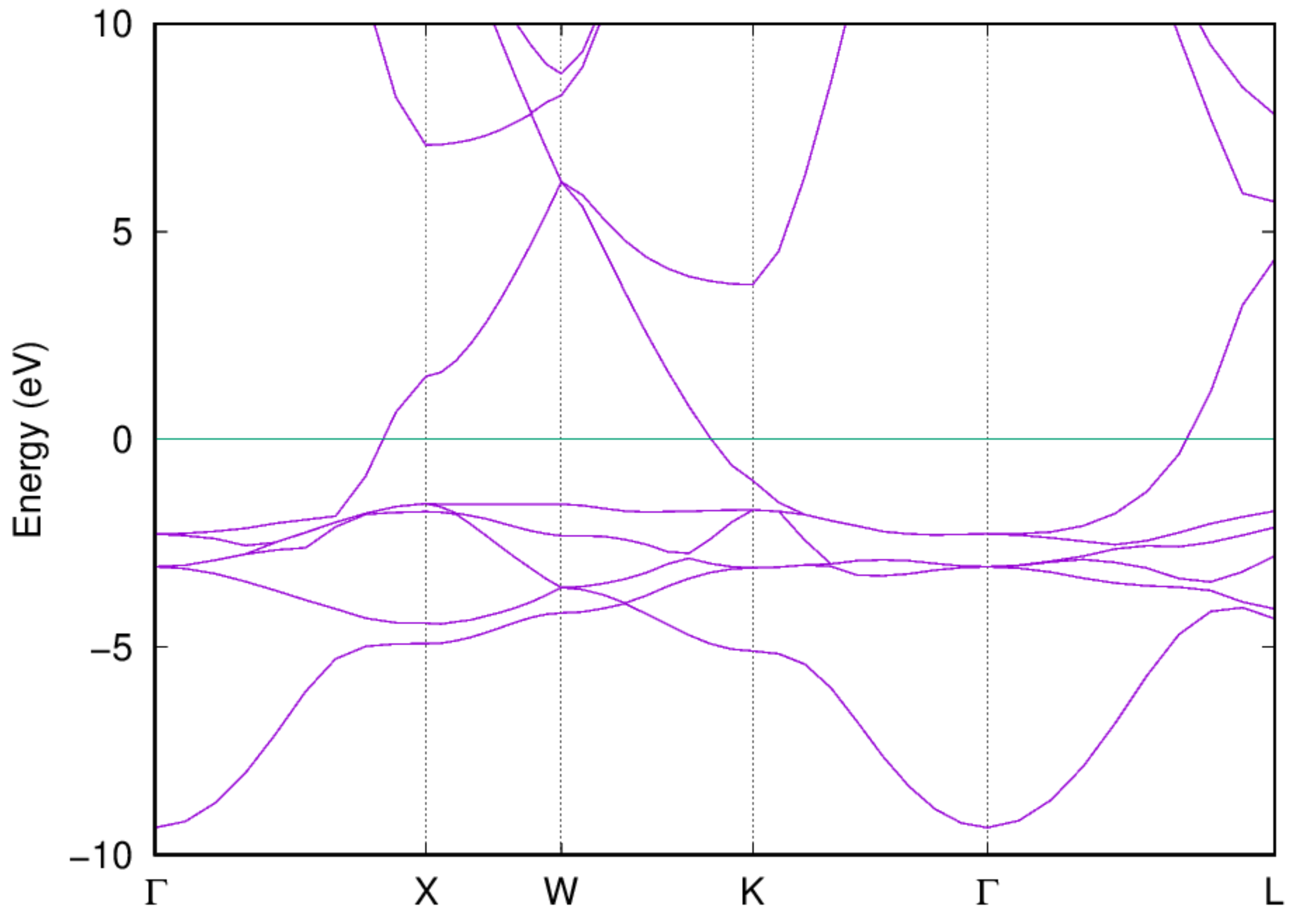
- You should also be able to get the electronic DOS via the following command:
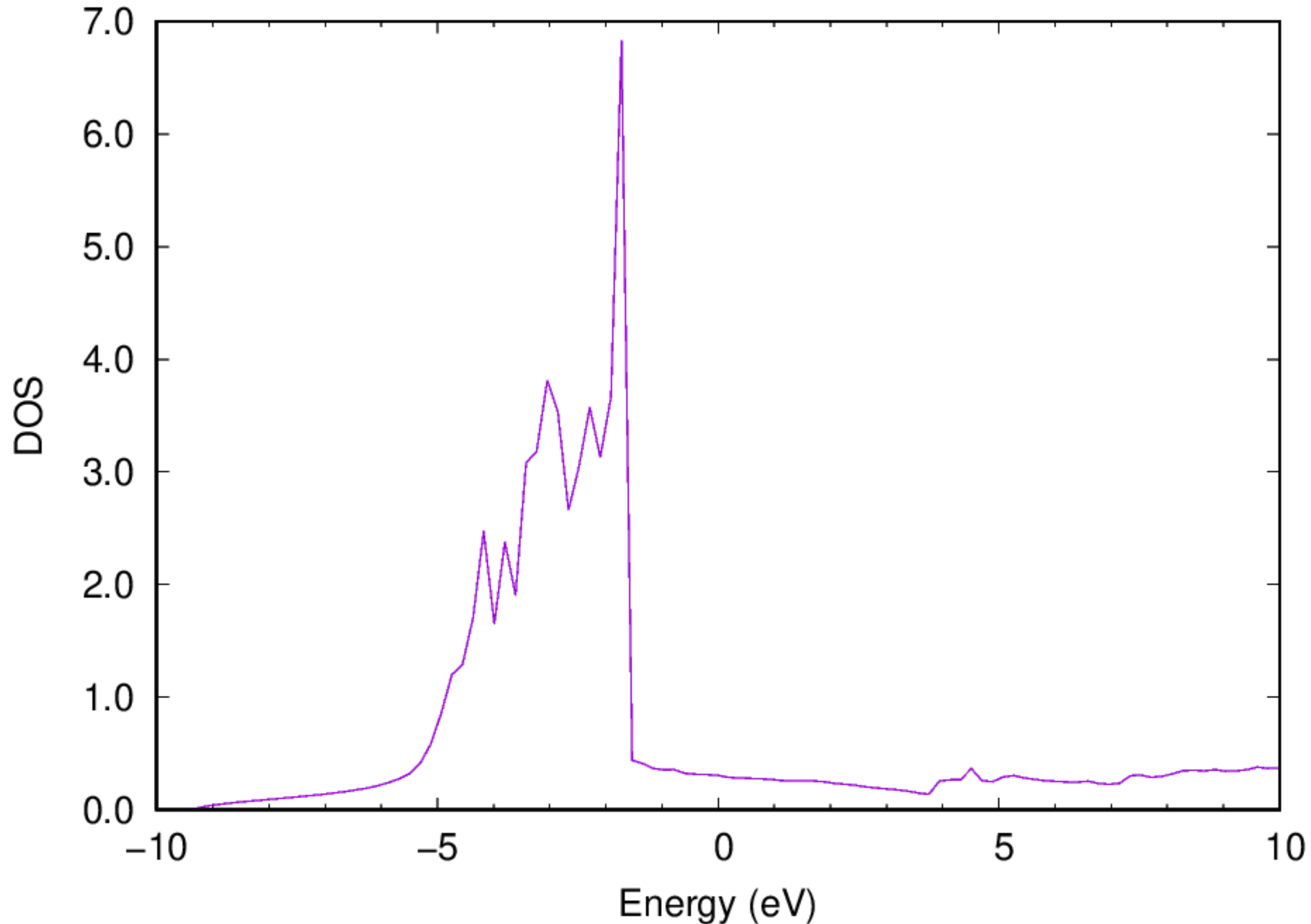
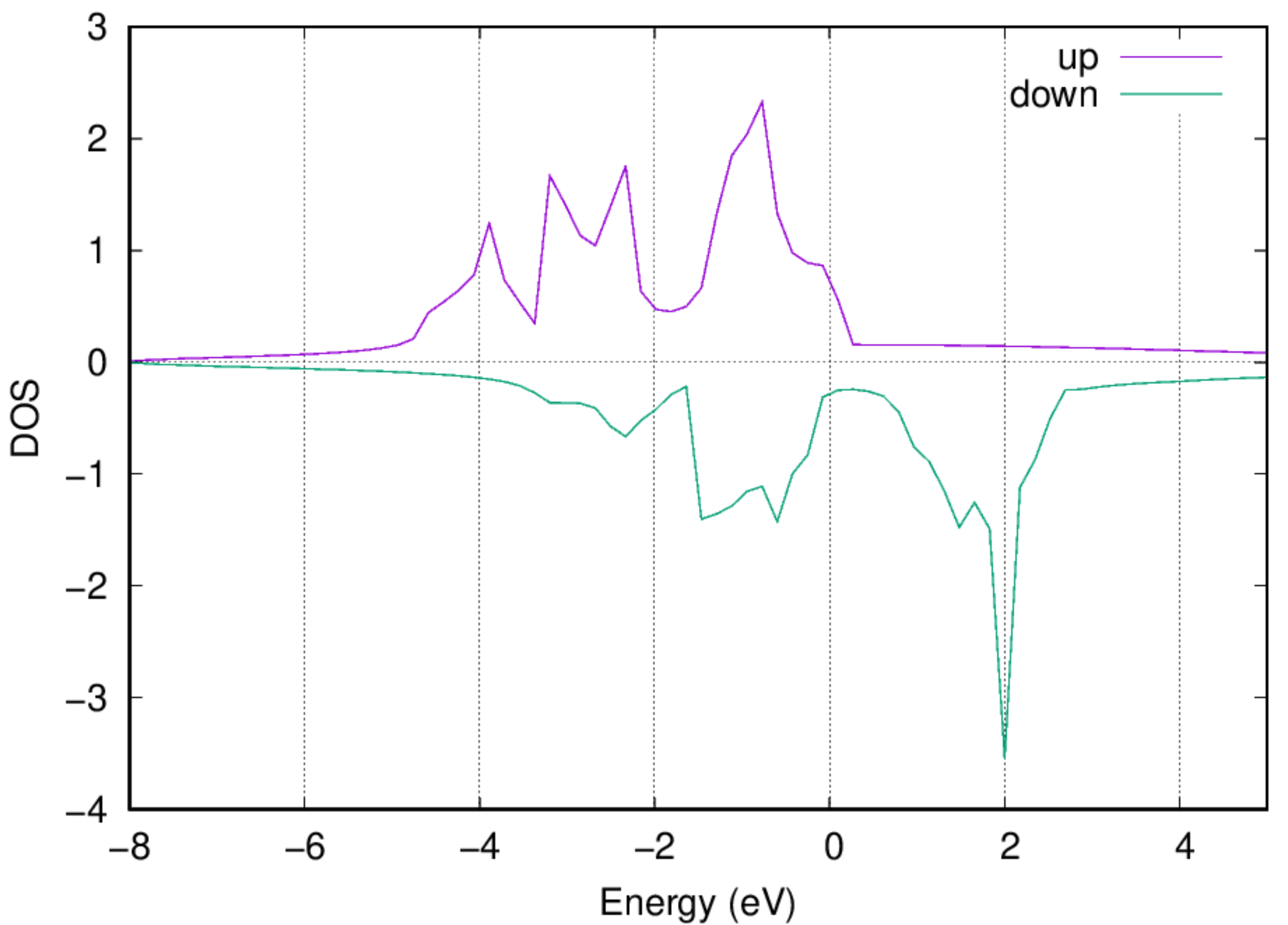
:::danger Note: The Fermi energy has been shifted to be zero. :::
2.3.2. BCC Fe¶
- Get into the directory
:::danger
Please read through all the main input files of vasp, especially INCARs and KPOINTSs.
:::
- Check the scripts
- Run the calculation
- Examine the band structure and DOS: Once the job is completed, you should be able to get the band structure via the following command:
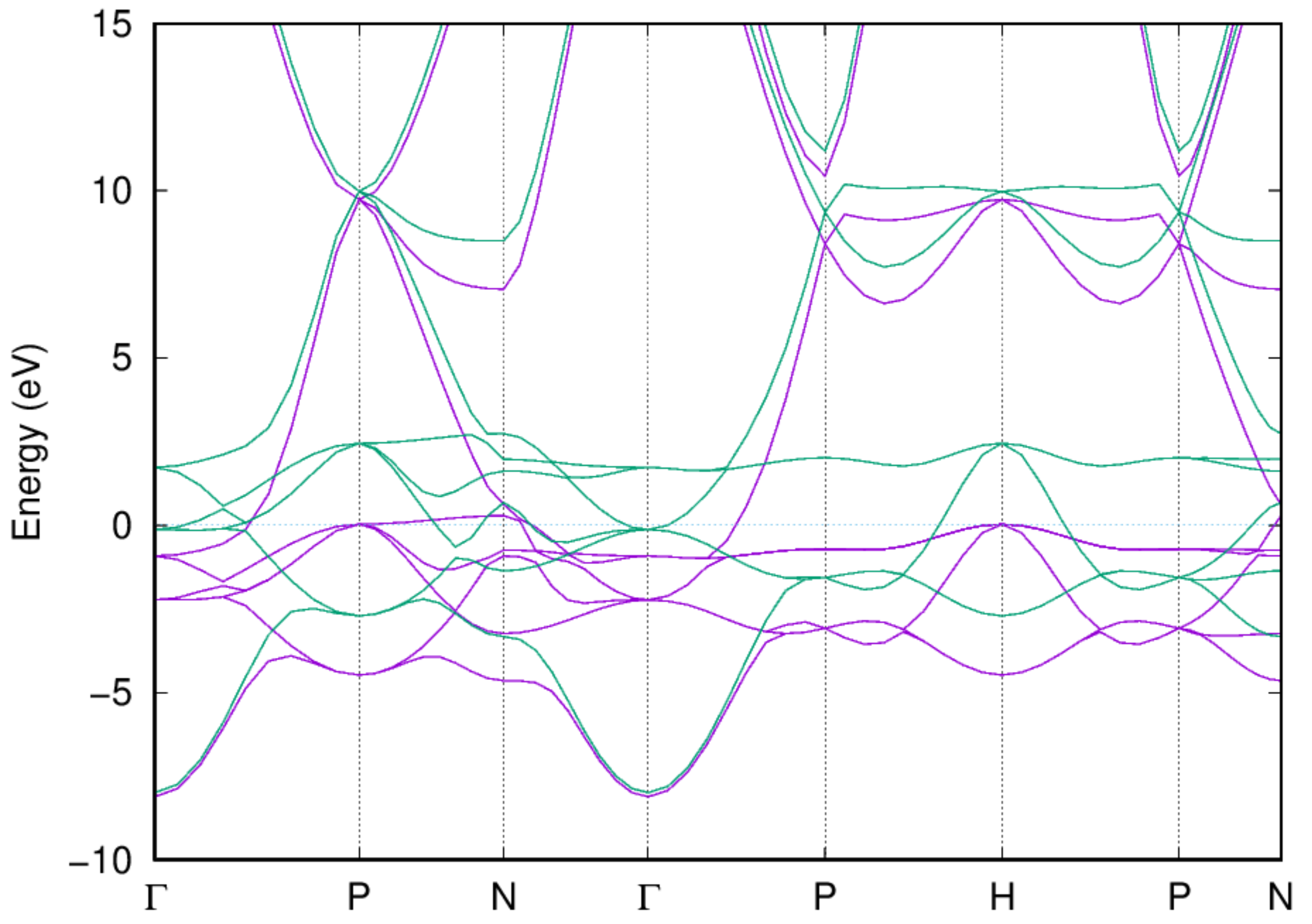
- You should also be able to get the electronic DOS via the following command:
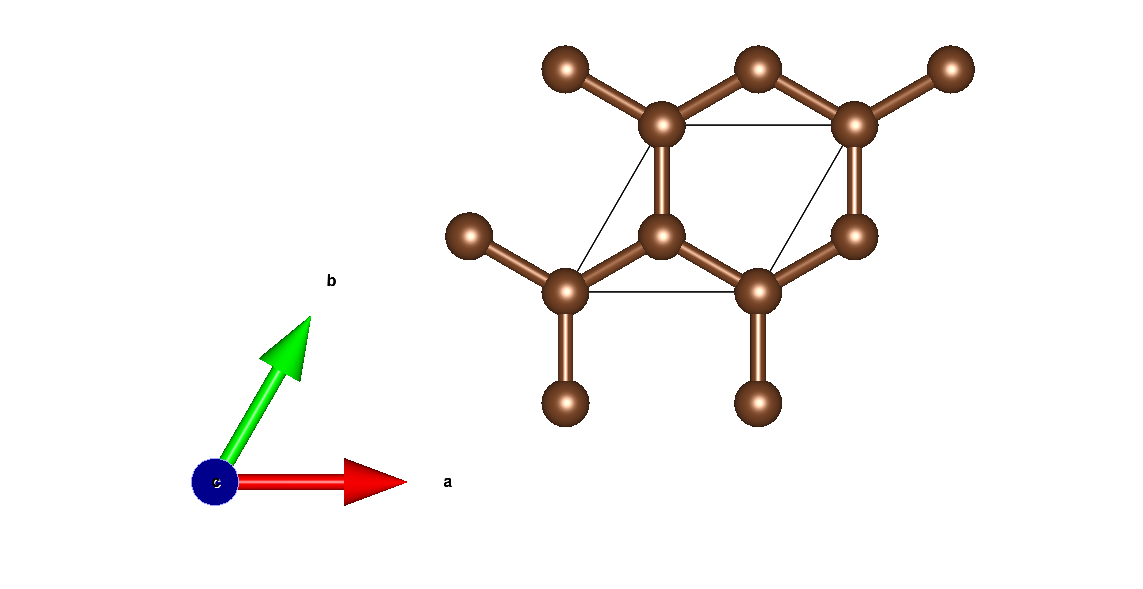
2.3.3. Graphene¶
- Graphene structure:
- Get into the directory
:::danger
Please read through all the main input files of vasp, especially INCARs and KPOINTSs.
:::
- Check the scripts
- Run the calculation
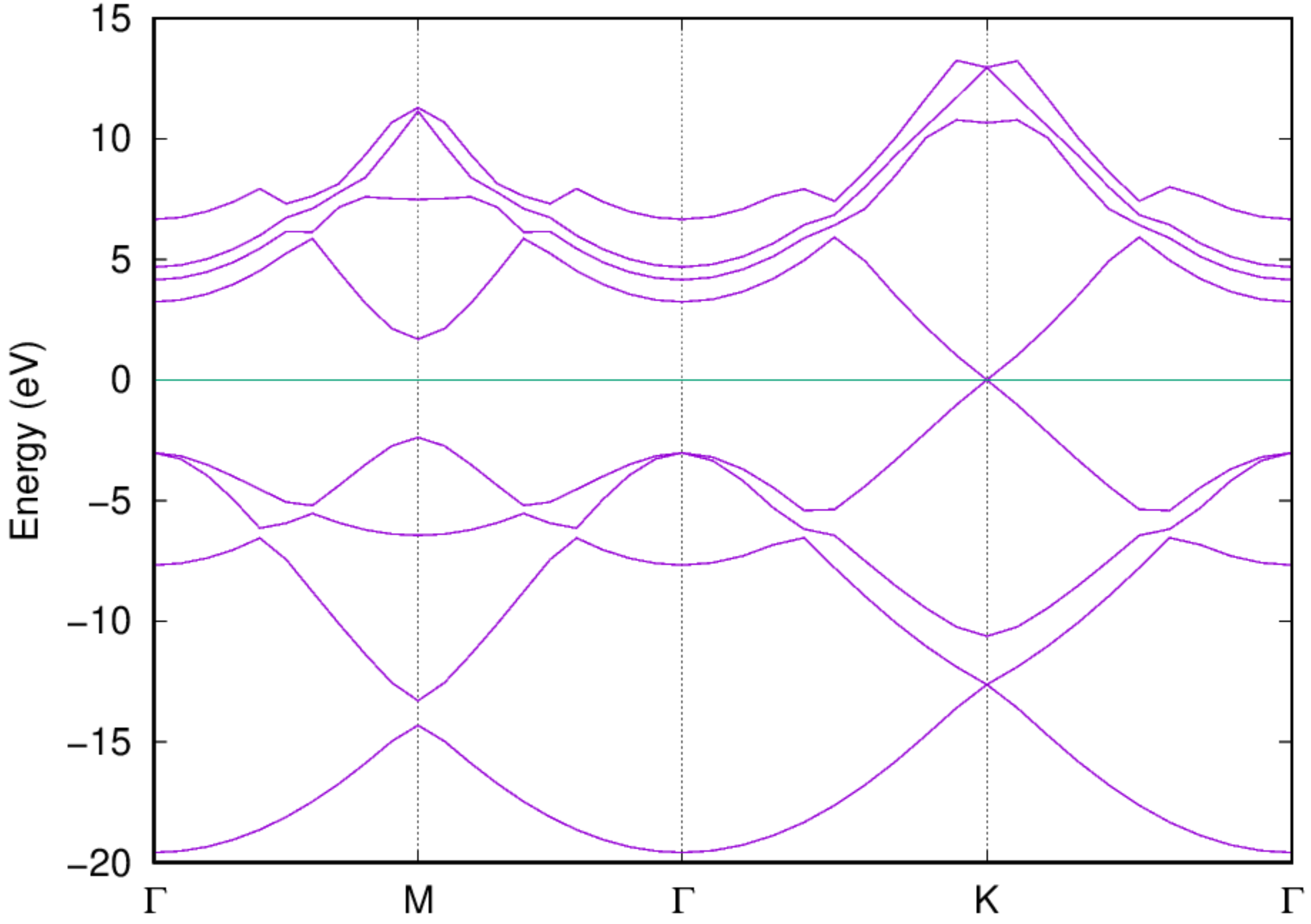
- Examine the band structure and DOS: Once the job is completed, you should be able to get the band structure via the following command:
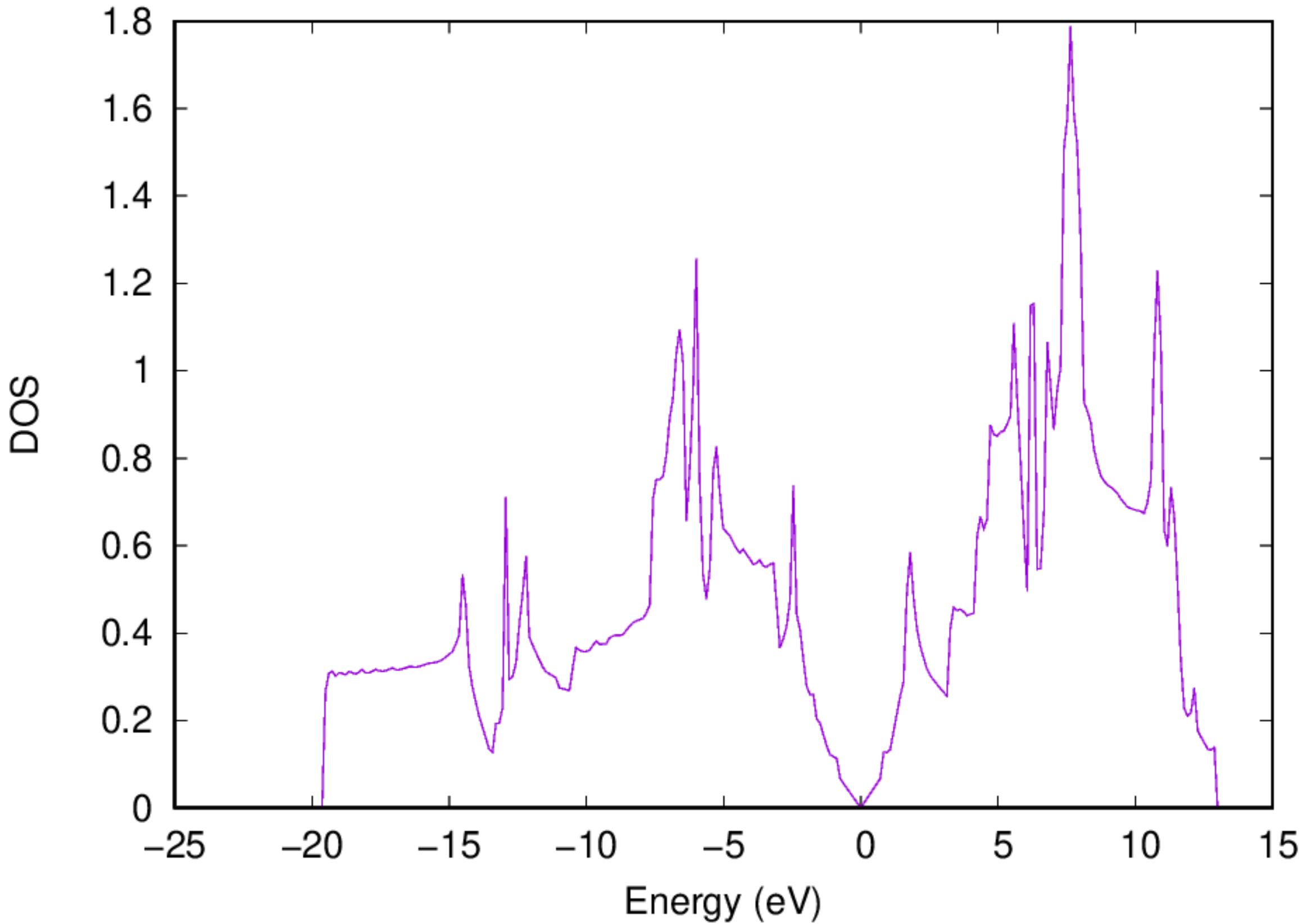
- You should also be able to get the electronic DOS via the following command:
2.3.4. Polyethylene (-CH\(_2\)CH\(_2\)-)¶
This example illustrates the calculation of band structure for polyethylene (-CH\(_2\)CH\(_2\)-) molecule chain (of infinite length):

It has periodicity along x direction, while free boundary (supercell) condition along the other two. Large vacuum along y and z are therefore introduced.
- Get into the directory
:::danger
Please read through all the main input files of vasp, especially INCARs and KPOINTSs.
:::
- Check the scripts
- Run the calculation
- Examine the band structure: Once the job is completed, you should be able to get the band structure via the following command:
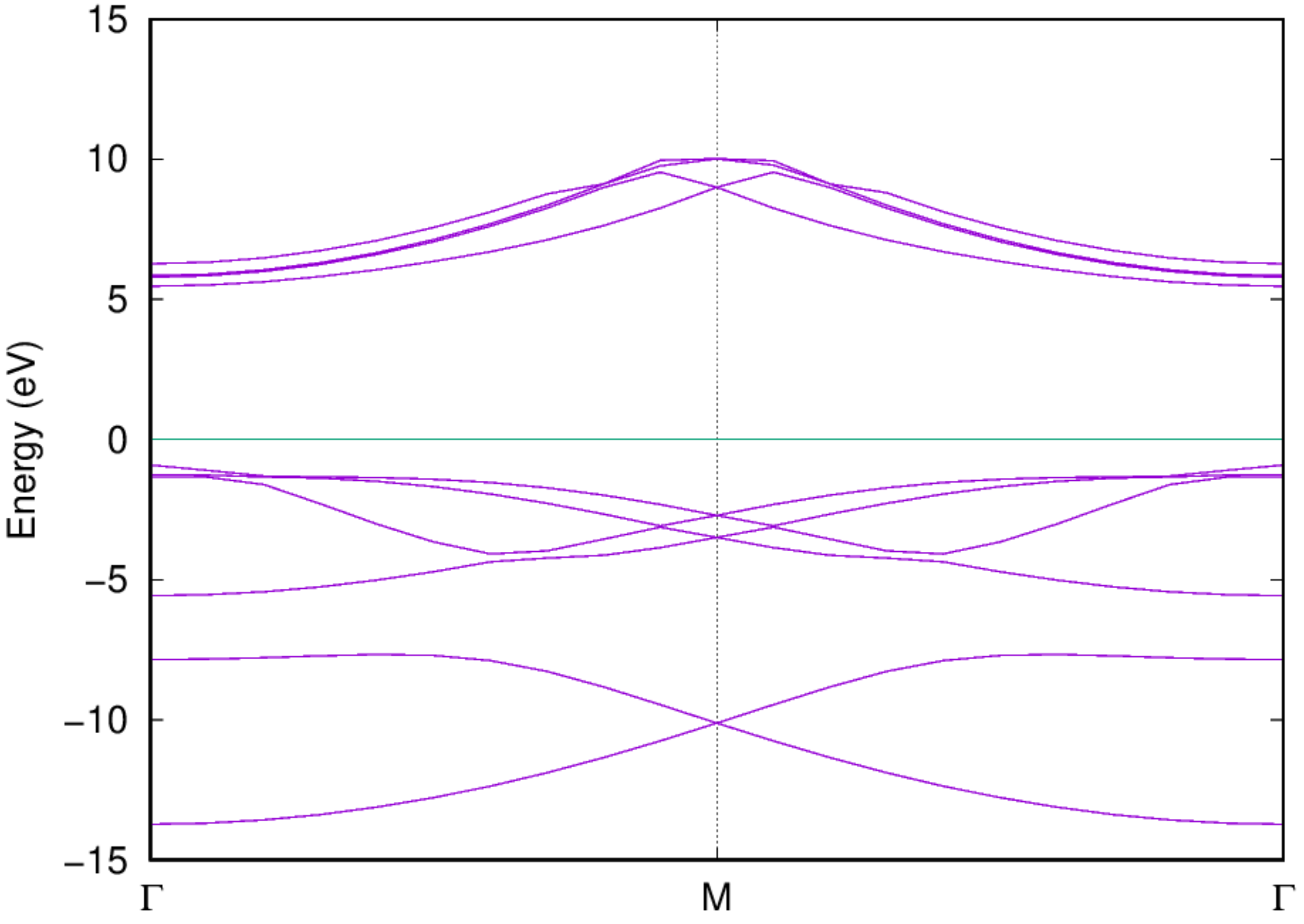
2.4. Bond length for H\(_2\) molecule¶
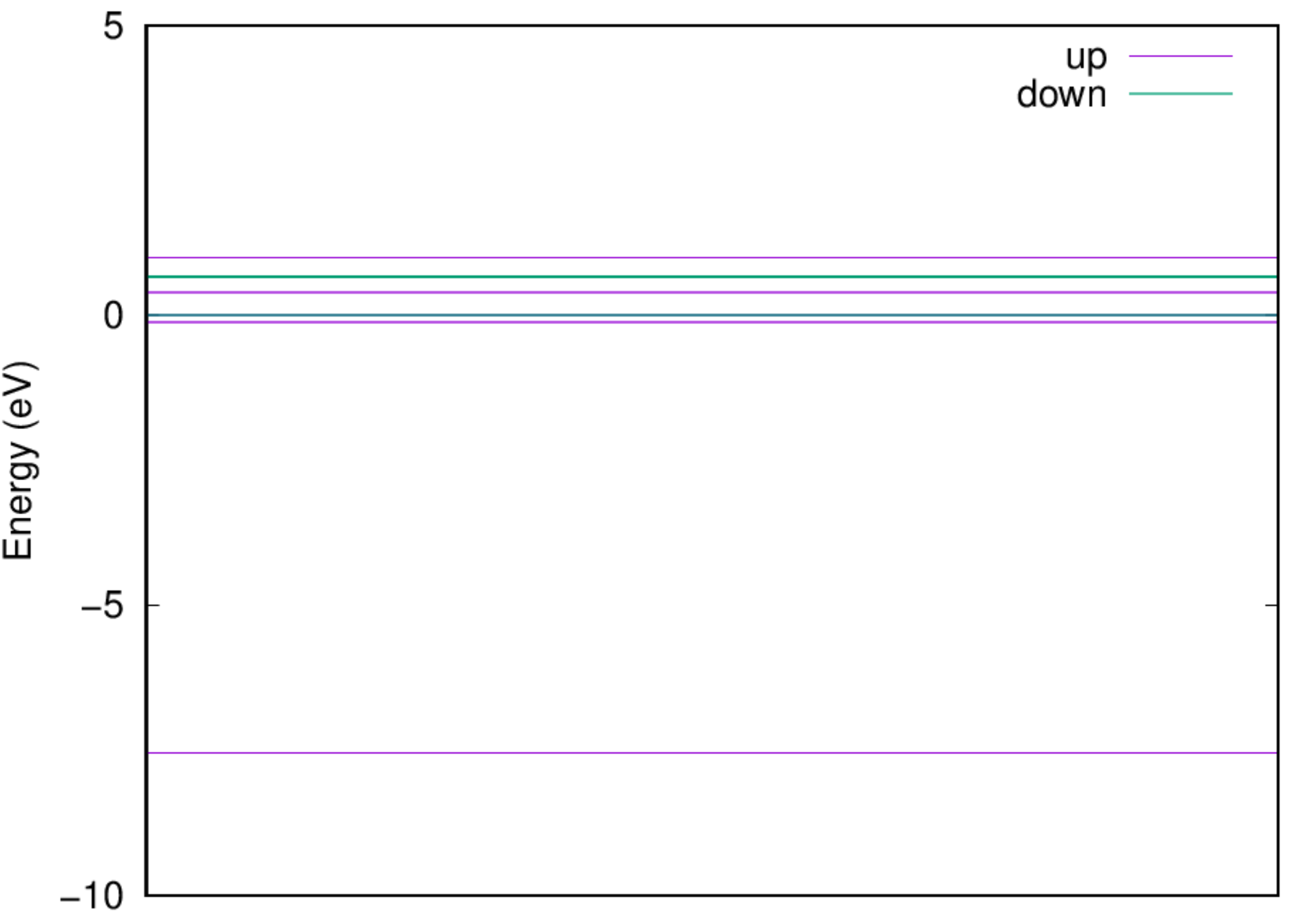
2.4.1. Ground state of H atom¶
- Get into the directory
- Check the scripts
- Run the calculation
:::danger Note: Since we are calculating a single isolated atom, a large supercell is used, and no periodicity is expected. Therefore we use only the \(\Gamma\) k-point. And since the supercell is large, many \(\mathbf{G}\) are within the cutoff radius and in turn the calculation will take some time. :::
- Examine the energy level: Once the job is completed, you should be able to get the energy level via the following command:
- You should also be able to get the total energy of \(H\) atom via the following command:
:::danger NOTE: there are exactly two spaces between 'free' and 'energy'. :::
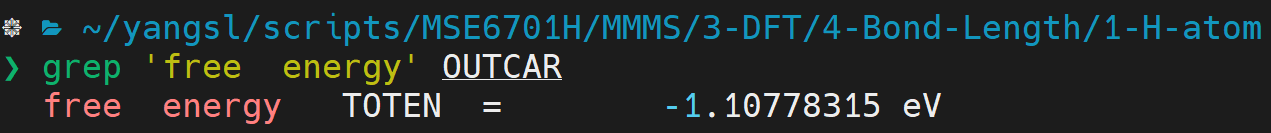
- Let's denote the ground state energy of a single \(H\) atom as \(E_H\).
2.4.2. Relaxation of \(H_2\) molecule¶
- Get into the directory
- Check the scripts
- Run the calculation
- Examine the energy level of \(H_2\): Once the job is completed, you should be able to get the energy level via the following command:

- You should also be able to get the total energy of \(H\) atom via the following command:
:::danger NOTE: there are exactly two spaces between 'free' and 'energy'. :::

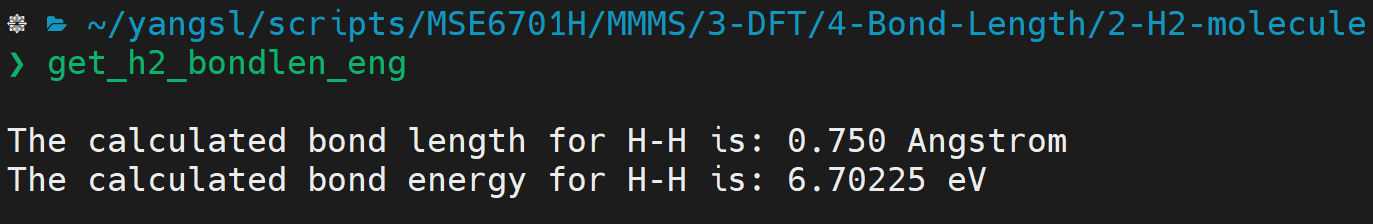
- Let's denote the ground state energy of \(H_2\) as \(E_{H_2}\), the \(H-H\) bond energy can be derived as:
- Bond length and energy of \(H_2\): The bond length and bond energy can be obtained from the final geometry:
:::info The experimental bond length and bond energy are about \(0.74\) Å and \(4.52\) eV, respectively. :::
2.5. Surface energy calculation¶
In this example, we will illustrate the calculation of surface energy for Cu(001). Generally, the surface energy of a clean metal surface can be calculated by three steps:
-
Static calculation of a bulk crystal, serving as a reference; here we will skip it, as it has been calculated in determining the lattice constant of Cu. Assuming the calculated bulk energy is \(E_{bulk}\) (per atom).
-
Construct a surface model, and make a static calculation, assuming the obtained total energy is \(E_{unrelaxed}\) and the total number of atoms in the surface model (slab) is \(N\).
-
Fix bottom three layers of the surface model, and allow the other atoms to relax freely. Make a relaxation run, and then a static run with the relaxed configuration (
mv CONTCAR POSCARand run a static calculation.) Assuming the final energy of the relaxed surface model is \(E_{relaxed}\).
The surface energy of the model should be given by:
$$ \gamma = \frac{E_{unrelaxed}-N E_{bulk}}{2A} + \frac{E_{relaxed}-E_{unrelaxed}}{A} $$
The as-cleaved surface model has two surfaces, and therefore a factor of 2 in the first term of the above equation; while the relaxation is performed only on the top surface, hence no factor of 2 in the second term.
FCC-Cu(001) 1x1x7+12 Ang vacuum structure:
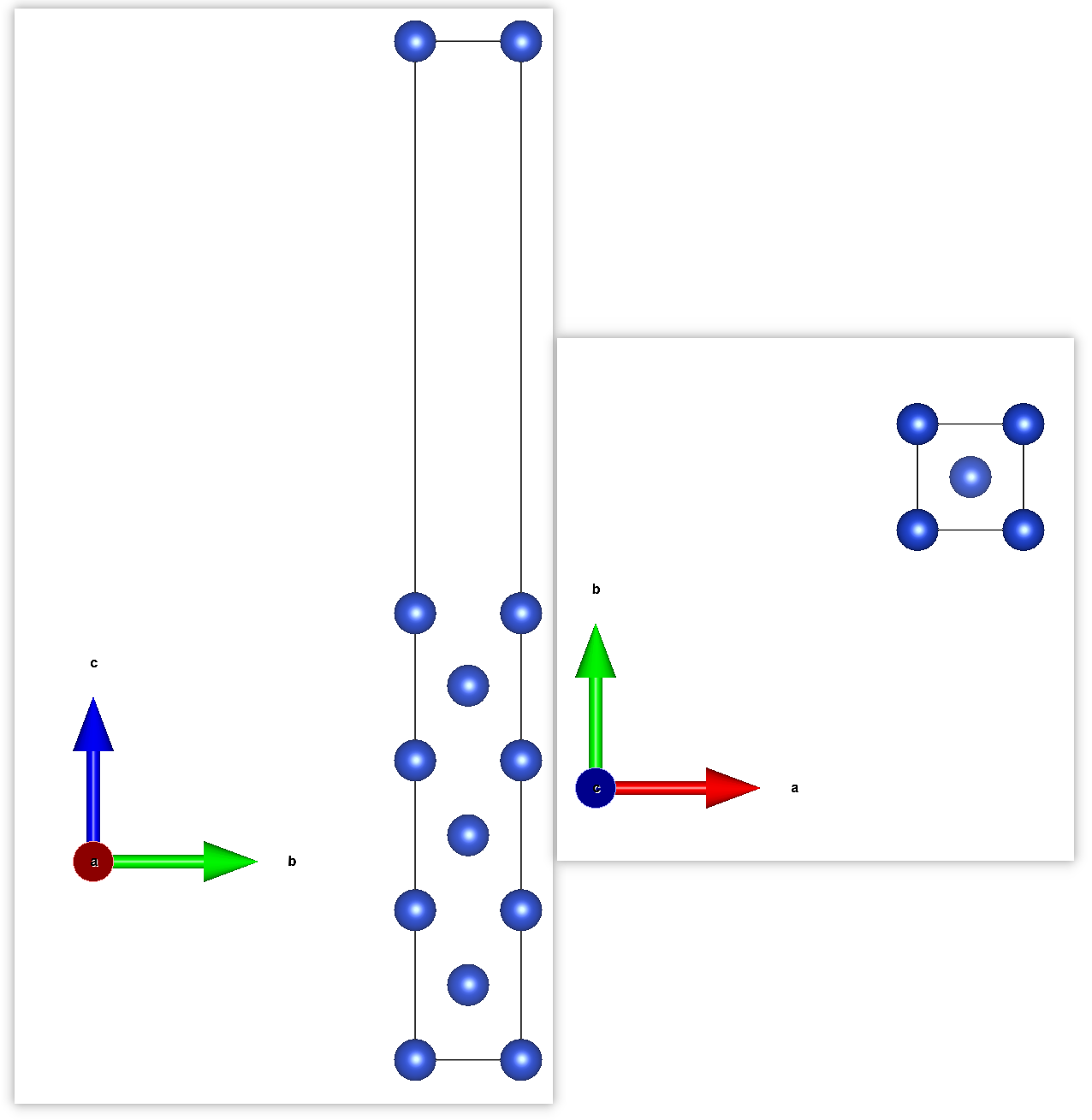
- Get into the directory
:::danger
Please read through all the main input files of vasp, especially INCARs and POSCARs.
:::
- Check the scripts
- Run the calculation
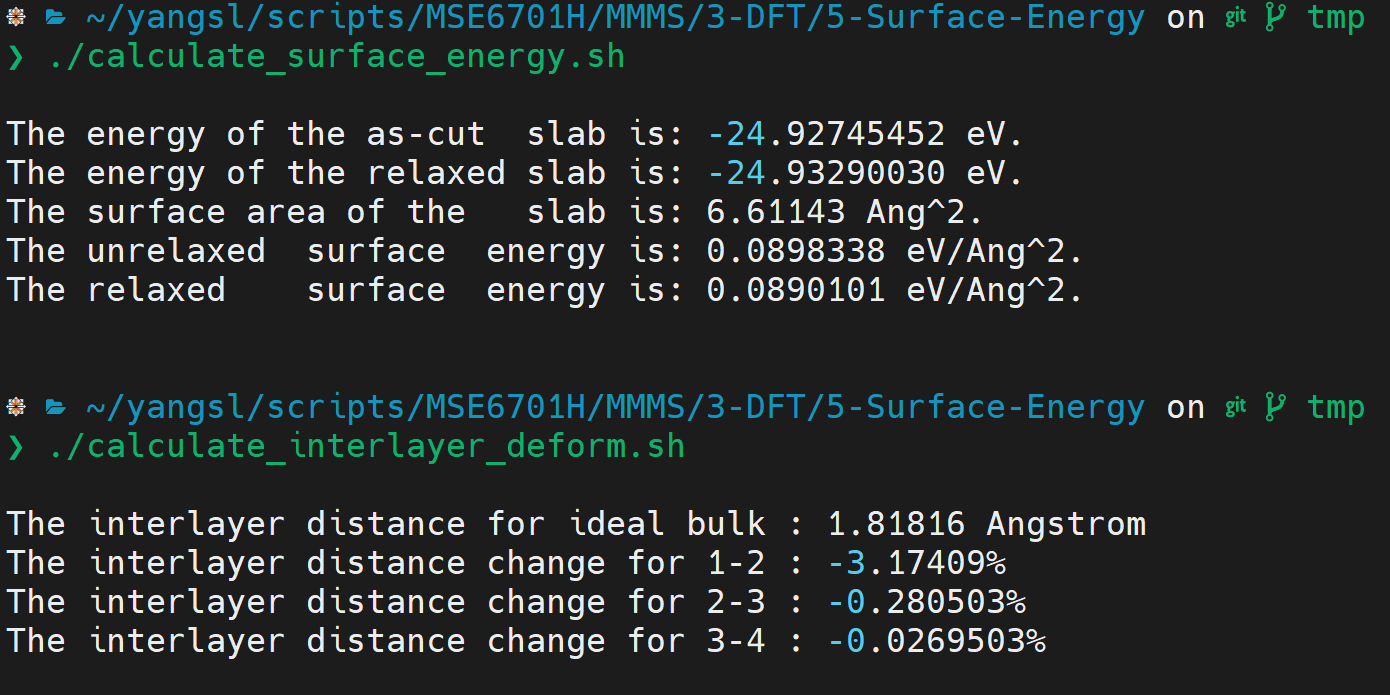
- Work out the surface energy and interlayer distance change: Once the calculations are done, you can check the
OUTCARto confirm the force on each atom, and check the atomic position for the atoms, so as to work out the change in interlayer distance for the surface model. Alternatively, you can also use the script provided to find out the surface energy and the relative change in interlayer distances:
2.6. Vacancy formation energy¶
The vancancy formation energy is defined as the excess energy when creating a vacancy in an otherwise ideal crystal:
where \(E_{vac}\) is the total energy of the supercell with a vacancy, while \(E_{bulk}\) is the total energy of the supercell without any vacancy.
Theoretically speaking, \(E_{bulk}\) can also be obtained from the calculation for a pritimive bulk cell. In practice, however, due to the difference in smearing method, k-mesh size, etc., there might be some difference in the total energy between a bulk supercell and a primitive one. This might translate into large uncertainty in the obtained vacancy formation. It is therefore advised to perform two separate calculations:
-
Static calculation for supercell without vacancy;
-
Relaxation of the supercell with a vacancy, and then continue with a static calculation.
The energies obtained from these calculations can then be used to calculate the vacancy formation energy.
2.6.1. Static calculation for supercell without vacancy¶
- Get into the directory
cd ../6-Vacancy-formation-energy/1-without-vacancy
# or
cd ~/MSE6701H/3-DFT/6-Vacancy-formation-energy/1-without-vacancy
:::danger
Please read through all the main input files of vasp, especially INCAR、POSCAR and KPOINTS.
:::
- Check the scripts
- Run the calculation
- The total energy can be find by:
:::danger NOTE: there are exactly two spaces between 'free' and 'energy'. :::

2.6.2. Relaxation and then static calculation for supercell with a vacancy¶
- Get into the directory
:::danger
Please read through all the main input files of vasp, especially INCARs、POSCAR and KPOINTS.
:::
- Check the scripts
- Run the calculation
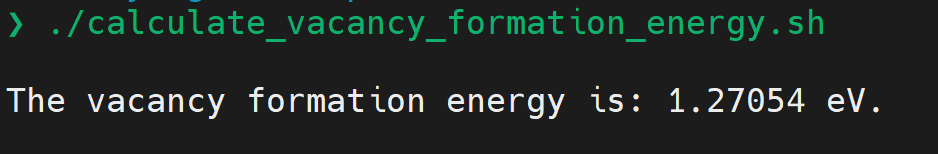
- Work out the vacancy formation energy
3. DFT Course Project¶
:::info This will be your second course project. The due is: Jan 22th, 2025. :::
You can follow the examples of 1-single-point-fcc-Cu and 2-lattice-constant-fcc-Cu to finish your homework. The pseudopotential file needed can be obtained by: